


DeepConformer is a model developed by us using a Deep Generative AI algorithm for predicting the dynamic structures of proteins. This model can be used to predict the dynamic conformations of 3D protein structures directly from protein sequences. Our algorithm for dynamic conformations can quickly obtain the dynamic properties of proteins, and can be directly used for multiple tasks such as protein design based on structure–activity relations as well as the prediction and optimization of a protein’s physicochemical properties.

The protein generation platform developed by us allows us to leverage the powerful modeling capability of Deep Generative AI to examine the evolutionary patterns and hidden relationships of protein sequences, so as to provide comprehensive solutions for drug design, enzyme engineering, etc. By generating diverse sequence variants with the physical properties of natural proteins, this platform is able to lay a solid foundation for drug development, bioengineering, and so on in the future.

We use a variety of methods for the simulation of molecular dynamics to capture the dynamic properties of protein molecules and explore their active sites and functional mechanisms. By establishing connections between protein conformations and their functional mechanisms, we can facilitate the design and optimization of biomolecules. The simulation of protein molecules is able to play a role in multiple applications such as providing or helping generate protein conformations, studying the active sites of proteins and their interactions with other molecules, calculating protein affinity, and predicting protein stability.

The fragment antigen-binding region (Fab region) is a region on an antibody that binds to antigens. It plays an essential role in immune responses. Utilizing publicly available Fab sequences and structural data, we developed an end-to-end structure prediction model that can precisely predict the constant and variable regions (CDR Loops) of an antibody’s Fab based on sequence information. The model allows researchers to comprehensively understand an antibody's overall structure and function, and offers insights into the antibody-antigen interaction mechanisms. It can be used to design therapeutic antibodies, develop vaccines, and modify antibody properties.

RNA as a tool for precisely regulating gene expression and protein synthesis is very valuable in treating diseases and bioengineering. Our RNA design platform uses an efficient algorithm based on an advanced Deep Generative AI model to decipher RNA sequences, structures and functions, so as to precisely design RNA molecules and help push the limits in drug development and biotechnology. The platform's sequence generating method can generate a high portion of active IRES sequences, which can be used for multiple applications such as the design of vectors for gene therapy and the creation of precision genetic circuits in synthetic biology.

We have AI platforms for predicting the secondary structures of aptamers, predicting the tertiary structures of nucleic acid-protein complexes, and predicting aptamer-protein interactions, as well as for exploring and validating the nucleic acid-protein binding mechanisms through molecular simulation.
Specifically, the platform for predicting aptamer-protein interactions uses a natural language model to capture the advanced features of protein sequences and aptamer sequences, so as to predict the possibility of aptamer-protein binding. This platform is trained using our own database for aptamer-protein interactions, and is highly sensitive to aptamer sequence mutations. It is designed to quickly screen for aptamer sequences that can bind to target proteins.

Modifying protein functions is a process of improving the specific functions of proteins by modifying their amino acid sequences or structures. We develop models for the prediction of functions by utilizing deep learning as well as internal and external datasets in connection with large-scale protein characterization and graph networks based on a pre-trained language model for proteins. Meanwhile, by utilizing our models for dynamic conformation and molecular simulation, we can predict, modify or improve protein functions such as catalytic activity, thermal stability, and affinity.
By utilizing multiple computational modules, we have developed a module for predicting protein properties that is highly specific and sensitive to sequence mutations. It can be used to help develop more efficient biocatalysts and industrial enzymes.
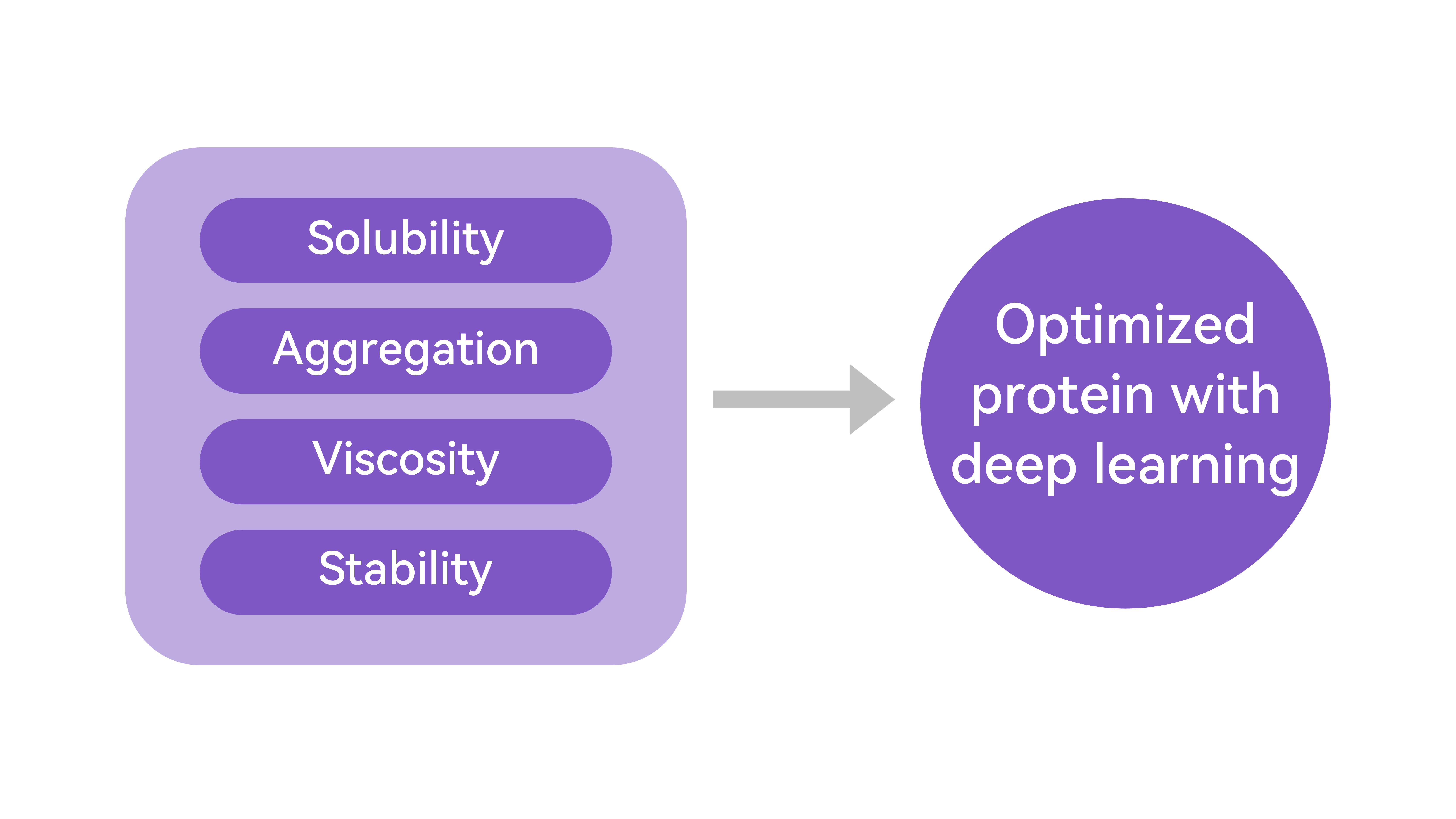
When it comes to drug design, the key is to screen and assess the metrics for druggability. The important attributes of protein druggability include, but are not limited to, their solubility, aggregation rate, half-life, stability, toxicity, and immunogenicity. By leveraging a multimodal fusion deep learning network, we can effectively predict protein properties such as their solubility, stability, and aggregation rate based on their sequence and structural information. This has significantly increased the accuracy and robustness in predicting various attributes related to druggability.

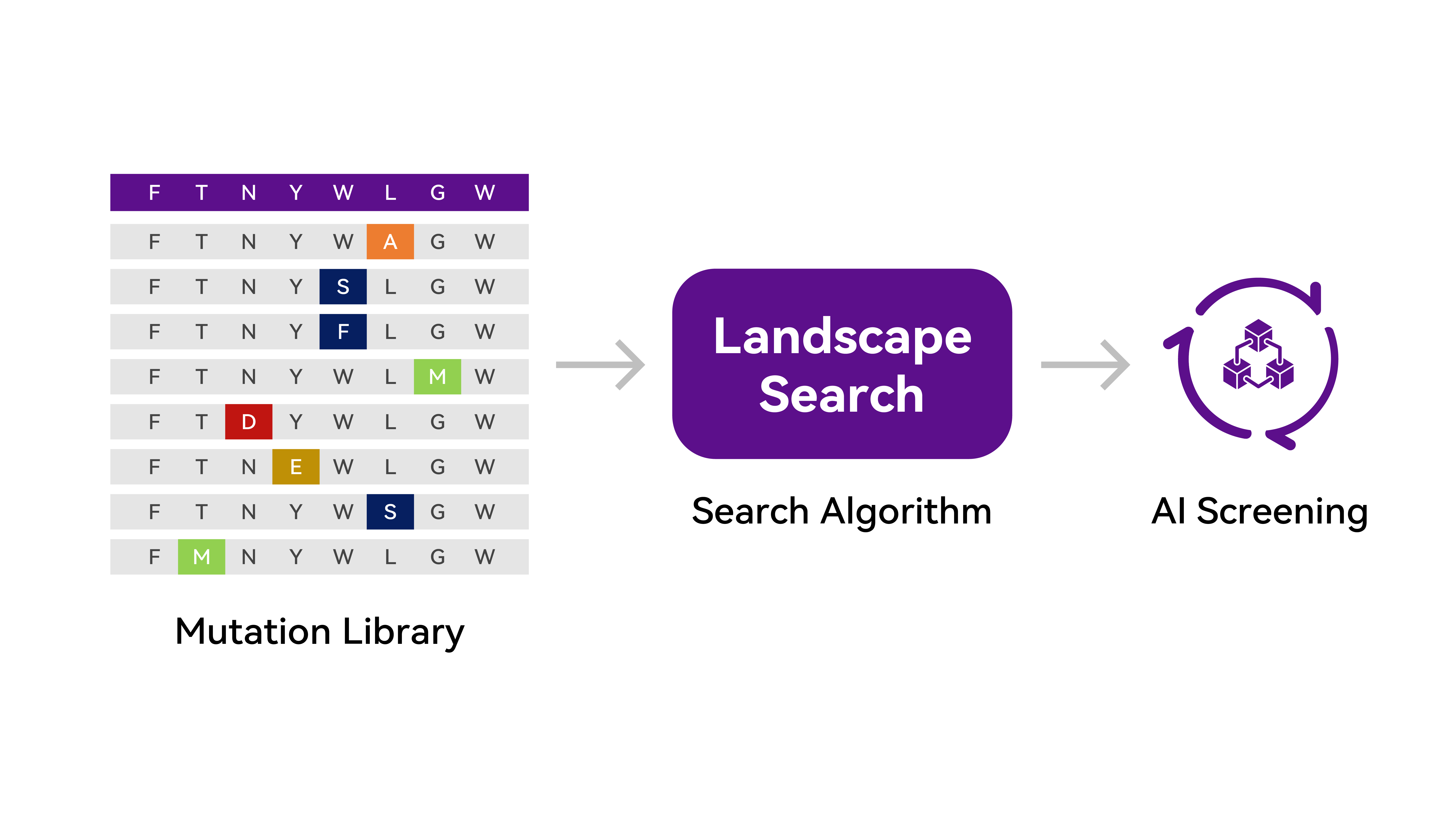
For specific targets and different tasks, we are able to do high-throughput generation, prediction and optimization by leveraging our unique capabilities for molecular simulation and the prediction of dynamic conformations. Based on our advanced Multi-Objective Optimization algorithm, we can probe into the complexity of protein structural topology at a single site, at multiple sites or even for exhaustive mutations, to help improve the key metrics for optimizing protein functions and modifying enzyme activity.
Molecular evolution and phylogenetics are key research methods for revealing the origins of life, understanding biodiversity, and developing novel therapies. The evolution of biological sequences is driven by mechanisms such as mutation, recombination, and selection. These mechanisms have generated rich genetic diversity at the molecular level. By analyzing the sequence information of biomolecules, we can trace the conservation and variability of genes and proteins with respect to their functions and structures, reveal their evolutionary relations, and predict their future evolution. Molecular evolution and phylogenetics are widely used for disease research, vaccine development, biotechnology, etc.
